Cell culture may seem straightforward once you’ve mastered the basics — preparing flasks, feeding cells, passaging at confluency — but true excellence lies in the fine details. Small deviations in key parameters can introduce major inconsistencies into your experiments, leading to unexpected variability, loss of cell phenotype, or even assay failure.
It’s not just about technique: the materials you use, such as your basal medium (like DMEM, RPMI, or MEM) and supplements like fetal bovine serum (FBS), play critical roles in influencing cell behaviour. Variations in media composition, serum batch quality, or culture conditions can significantly impact growth rates, viability, morphology, and experimental reproducibility. Even when using trusted brands, monitoring and optimising your culture environment remains essential.
Whether you’re working with primary cells, mesenchymal stem cells, or immortalised cell lines, understanding and tracking specific culture metrics ensures cell health, improves reproducibility, and strengthens your experimental outcomes.
In this article, we highlight seven critical numbers every researcher should monitor — and explain why they matter for achieving consistent, high-quality results.
1. Passage Number / Population Doubling
The passage number reflects how many times a population of cells has been subcultured (split and reseeded into new vessels) since its original isolation or revival from cryostorage. Each passage marks a round of cell expansion and propagation.
Although passage number is often treated as a routine metric, it plays a critical role in determining cell quality, performance, and experimental reproducibility. Over time, cells accumulate molecular and epigenetic changes during in vitro culture. These changes can lead to significant shifts in behaviour, such as altered gene expression, slower proliferation, reduced viability, or unwanted differentiation — even when cell morphology remains unchanged.
Why Do We Need to Monitor and Record Cell Passage Number?
Monitoring and recording passage number ensures:
Consistency across experiments: Using cells at different passage numbers can introduce variability into your data.
Predictable behaviour: Cells behave more consistently within a defined passage window. Beyond that, they may show signs of senescence or genetic drift.
Compliance and traceability: In GMP workflows or publication-quality research, full traceability of cell handling is expected — and that includes passage history.
Decision-making: Knowing the passage number helps researchers decide when to create new working stocks, discard aging cultures, or troubleshoot changes in phenotype.
Failing to track passage number can result in irreproducible results, misleading conclusions, and wasted resources — especially in long-term studies or preclinical development pipelines.
What Constitutes a Passage?
In routine cell culture, each subculture or reseeding is considered one passage.
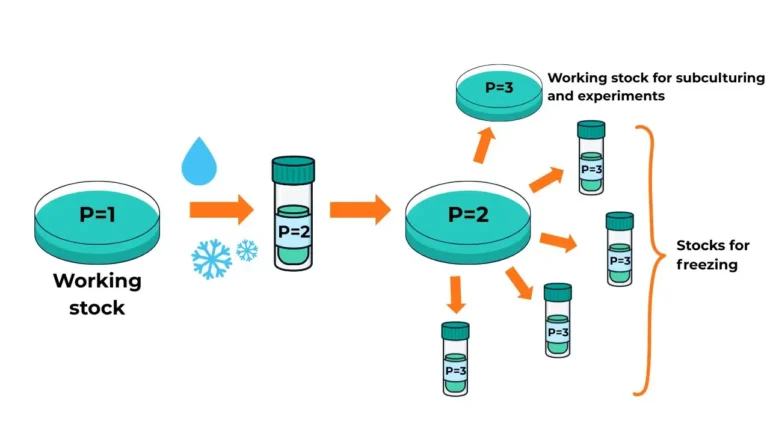
However, a common question arises: Does thawing frozen cells count as a new passage? The answer is no — passage number should increase upon reseeding, not upon freezing.
Example: If you have a working cell line at passage 1 (P1), and you trypsinise and freeze these cells, you label the vial as P2. When thawed, they remain P2 until you subculture them again, at which point they become P3.
Figure 1: Determination of Passage Number from Freezing Down Stock to Subsequent Subcultures
Why Passage Number Alone Isn’t Enough
Passage number does not account for split ratios or seeding densities, and therefore provides limited insight into how many times the cells have actually divided.
Consider this scenario:
Researcher A splits a culture at P10 using a 1:4 ratio.
Researcher B splits the same culture at P10 using a 1:10 ratio.
Both label the next flask as P11.
However, the cells split 1:10 must proliferate more to reach confluence, meaning they undergo more population doublings (PDs). Over time, this can result in cells that are genetically and functionally different, even though their passage numbers are identical.
Why PD Tracking Matters
More accurate: Reflects true replication history, especially when split ratios vary.
Informs senescence thresholds: Particularly important for primary cells and MSCs, where function declines with excessive replication.
Enhances comparability: Useful when comparing results across different donors, media formulations, or labs.
Supports translational research: Essential for regulatory documentation and reproducibility in clinical-grade workflows.
Monitoring PD in tandem with passage number and doubling time provides a multi-dimensional view of culture health, enabling better decision-making, especially in long-term or translational studies.
2. Cell Seeding Density
Cell seeding density — the number of cells plated per unit area (for adherent cells) or per unit volume (for suspension cells) — is a fundamental parameter in cell culture. It directly influences growth rate, morphology, gene expression, and the outcome of downstream assays.
Incorrect seeding can lead to:
Over-confluency, causing contact inhibition, altered signalling, or early senescence
Low density, which can stress cells, delay growth, or alter behaviour
Experimental variability, especially in assays dependent on cell–cell interactions, secreted factors, or differentiation cues
Maintaining an appropriate seeding density ensures:
Uniform cell distribution and attachment (for adherent cells)
Optimal nutrient and oxygen access
Consistency across replicates and time points
Maintenance of key cellular functions and phenotypes
Typical Ranges
Cell Type
Recommended Seeding Density
Adherent cells
5,000–50,000 cells/cm²
Suspension cells
2 × 104 to 5 × 105 cells/mL
These are only general ranges. Optimal densities vary based on the cell type, assay type, culture vessel size, and experimental goals.
Pro Tip: It’s recommended to seed more cells immediately after thawing than during routine passaging, as post-thaw cell viability is typically lower.
Factors That Influence Cell Seeding Density
Cell density requirements differ widely depending on:
Cell type – Some cells (e.g., primary neurons or endothelial cells) need tight cell–cell contact to survive, while others (e.g., fibroblasts) tolerate low-density seeding.
Consult published literature and perform pilot experiments to determine the most appropriate seeding density for your specific system. What works for one lab or cell line may not apply to another.
How to Calculate Seeding Density
Here’s a quick guide to help you calculate the correct seeding density for your experiment:
Determine the desired seeding density — e.g., X cells/cm²
Count your cells using a hemocytometer or automated counter
Calculate total cells needed by multiplying X with the total surface area of your vessel
Centrifuge your cell suspension and resuspend the pellet in a smaller known volume (e.g., 1 mL) — call this volume Y
Calculate the volume needed to reach the desired seeding density using:
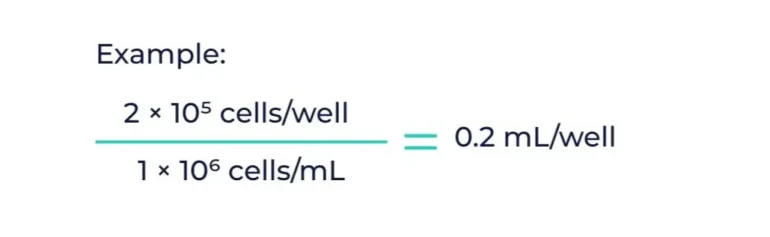
Example: You need 2 × 10⁵ cells/well in a 6-well plate. If you have 1 × 10⁶ cells/mL, you would need 0.2 mL per well.
3. Confluency Percentage
Cell confluency refers to the percentage of a culture surface covered by adherent cells. It is a quick and essential visual indicator of cell proliferation and overall culture health.
More than just a routine metric, confluency is used to guide decisions across the entire cell culture workflow—from passaging and cryopreservation to experimental treatments and differentiation. Importantly, cell behaviour, gene expression, and responsiveness to external stimuli are all highly influenced by confluency levels.
What Is Cell Confluency?
Confluency is not a direct count of cells, but rather a visual estimate of surface coverage. For example, a 70% confluent flask has 70% of its surface area occupied by adherent cells.
Used correctly, confluency helps determine:
When to split or passage cells
The best time for transfection or drug treatment
Whether the culture is suitable for downstream applications like differentiation, reprogramming, or harvesting
Figure 2: A schematic showing 4 fields of view under a microscope of cells depicted at 20%, 50%, 80% and 100% confluence. Reference: Cell culture basics and protocol
The Risks of Over-Confluency
Failure to passage at the right time can lead to:
Nutrient depletion and metabolic stress
Cells entering growth arrest, senescence, or apoptosis
Accumulation of toxic cellular debris from dying cells
Altered morphology and gene expression that misrepresent in vivo conditions
Higher risk of contamination, particularly fungal or bacterial
Why Accurate Estimation Is Critical
Despite its importance, confluency is still often estimated by eye under a microscope. This “guesstimation” is subjective and can vary between users—even for the same person on different days. Estimating between 60–90% confluency, the most critical range for many procedures, is especially error-prone.
Accurate and consistent estimation of confluency supports:
Reproducibility: Ensures cells are in the same physiological state across experiments
Efficiency: Avoids wasted reagents and failed workflows due to mistimed actions
Experimental validity: Ensures transfection and drug treatments are performed under optimal growth conditions
Recommended Confluency Targets by Application
Application
Target Confluency
Rationale
Proliferation assays
60–80%
Ensures active growth
Transfection/Transduction
70–90%
Balances viability and optimal uptake
Differentiation
Varies by cell type
Some require high density; others require space for morphogenic changes
Cryopreservation
70–90%
Prepares cells at peak health for recovery post-thaw
Expansion post-thaw
50–70%
Prevents overcrowding and supports healthy regrowth
Pro Tips for Better Confluency Estimation
Use reference images for training and standardisation
Keep a photographic log of typical confluency stages
Use quantification tools like ImageJ or confluency analysis software when possible
Adopt automated imaging systems for objective monitoring
Strategies to Prevent Over-Confluency
Over-confluency is a preventable issue that can derail entire experiments. Use these proactive strategies:
Regular Monitoring: Check your cultures daily under a microscope and record observations. Unexpected growth spurts can happen quickly.
Optimise Seeding Density: Starting with too many cells can lead to rapid overgrowth. Tailor seeding density to your cell type, vessel size, and application.
Use Larger Culture Vessels: If frequent passaging becomes burdensome, scaling up to larger flasks or plates may reduce handling frequency.
Adjust Media Change Frequency: Fast-growing cells may deplete nutrients quickly. More frequent media changes can help sustain viability and delay confluence.
Optimise Culture Conditions: Ensure proper CO₂ levels, humidity, and temperature. Cells under stress may behave unpredictably, affecting growth dynamics.
Establish SOPs and Thresholds: Define clear confluency thresholds in your protocols—for example, “Passage cells when 80% confluent”—to improve consistency across the lab.
4. Viability Percentage
Cell viability is a key indicator of overall cell health and is critical for ensuring reliable and reproducible experimental outcomes. Whether you’re conducting transfections, drug assays, differentiation studies, or preparing cells for cryopreservation, maintaining a high viability percentage helps ensure consistent and physiologically relevant results.
Low viability can:
Distort assay outcomes due to interference from dead or dying cells
Trigger stress responses that alter gene and protein expression
Lower transfection or infection efficiency
Cause batch-to-batch inconsistencies that undermine reproducibility
Target Threshold
A healthy cell culture is characterised by 80-95%, and is generally recommended for most standard experiments.
FDA Guidance: For ex vivo gene-modified cells (e.g., CAR-T, MSCs), the FDA recommends a minimum viability of 70%. If lower, supporting data must show it doesn’t affect safety, efficacy, or delivery, as dead cells or debris may compromise the therapeutic outcome.
How to Assess Viability
Trypan Blue Exclusion: Simple, manual counting method using a hemocytometer or automated counter.
Fluorescent or colourimetric assays (e.g., MTT, Resazurin): Suitable for high-throughput screening.
The Role of FBS in Cell Viability
FBS is one of the most critical components in many culture media formulations. It provides essential growth factors, hormones, adhesion molecules, and nutrients that support cell attachment, proliferation, and survival.
Poor-quality or variable FBS can lead to unpredictable cell behaviour and low viability.
Contaminated or improperly heat-inactivated FBS may cause cytotoxic effects.
Batch-to-batch variability in FBS can create inconsistencies in experimental results unless the FBS is rigorously tested and validated.
Pro Tip – Always source FBS from reputable suppliers that offer:
Certified virus and mycoplasma-free products
Proven batch consistency with pre-testing options
Traceable origin and ethical collection practices
Common Causes of Low Viability
Suboptimal thawing or freeze–thaw cycles
Over-confluent cultures leading to nutrient depletion
Harsh enzymatic detachment
Contamination (e.g., mycoplasma, endotoxins)
Poor media quality or inappropriate supplements like unstable or inconsistent FBS
Tips to Improve Cell Viability
Use pre-warmed, complete media with high-quality FBS
Avoid over-handling or exposing cells to room temperature for prolonged periods
Optimise enzymatic detachment conditions
Maintain proper seeding density after thawing to encourage recovery
Perform routine QC (e.g., mycoplasma testing, media pH, osmolality checks)
High viability isn’t just about surviving—it’s about ensuring your cells are healthy, active, and behaving as expected. Trustworthy reagents like certified FBS form the backbone of a reliable culture system.
5. Mycoplasma Testing Frequency
Mycoplasma contamination is one of the most insidious and underdiagnosed problems in cell culture. Unlike bacterial or fungal contamination, mycoplasma does not cause turbidity in the media and often goes unnoticed—while silently wreaking havoc on experimental integrity.
These tiny, cell wall–less bacteria can:
Alter cell metabolism, proliferation, and gene expression
Interfere with transfection efficiency and cytokine responses
Compromise reproducibility and lead to false experimental conclusions
Go undetected for weeks or months, especially in shared incubators or labs
Recommended Testing Frequency
Situation
Testing Frequency
Routine cell culture maintenance
Every 4–6 weeks
Before critical experiments
At least 1 week prior
Before cryopreserving cell stocks
Mandatory
After thawing cells from storage
Within 3–5 days post-thaw
After receiving new cell lines
Immediately upon arrival
After antibiotic treatment or suspected contamination
Follow-up testing required
High-Risk Scenarios Needing Frequent Monitoring
Shared or core lab environments
Long-term culture experiments
Laboratories working with primary or stem cellsS
Labs using pooled or co-cultures
Facilities preparing cells for clinical or preclinical use
DNA staining (e.g., Hoechst 33258): Useful for microscopy-based detection
Enzymatic assays: Quick and simple for routine screening
Culture-based assays: Gold standard but time-consuming and not suitable for rapid decisions
Best Practices
Quarantine new cell lines until cleared
Maintain detailed records of all test results
Implement standard operating procedures for contamination response
Avoid using antibiotics long-term to mask contamination
Proactive, scheduled mycoplasma testing and frequent mycoplasma decontamination practice is a small investment of time that can save months of compromised work.
6. pH Range of Culture Medium
The pH of culture media plays a crucial role in influencing cell viability, metabolism, enzyme activity, and overall experimental reproducibility. Any deviation from the optimal pH range can lead to altered cellular behaviour, unreliable data, or even complete culture failure. Maintaining stable pH is especially critical for sensitive cell types, such as primary cells and stem cells.
The optimal pH range for mammalian cells is typically between 7.2 – 7.4. A pH below 7.2 is considered acidic, while a pH above 7.4 is regarded as alkaline.
What Do pH Changes in Cell Culture Media Indicate?
Acidic Conditions – Usually indicate overgrown cultures or possible microbial contamination. As cells metabolise nutrients, they release acidic byproducts such as lactic acid, which lowers the pH. This is particularly common in dense cultures or when media changes are infrequent.
Alkaline Conditions – May arise from insufficient CO₂ levels in the incubator or prolonged exposure of the culture dish to air. It can also occur when cells are not metabolically active, such as after cell death or in the early stages post-thaw.
pH Regulation Mechanisms
1. Phenol Red Indicator
Phenol red, a common pH indicator in cell culture media, shifts from pink (neutral) to orange or yellow (acidic), providing a visual cue to monitor media condition in real-time. A yellowing of the medium signals increased acidity and typically indicates the need for a media change, regardless of the scheduled feeding timeline.
Figure 3: Cell culture media are commonly supplemented with phenol red as a pH indicator. The colour of the medium reflects the pH level—pink indicates neutral pH, yellow signals acidic conditions, and purple suggests an alkaline environment. Reference: Culture Collection
Does Phenol Red Affect Cells?
In most cases, phenol red is harmless and useful for maintaining healthy cultures. However, certain sensitive cell lines or experimental setups may require phenol red-free media. This is because phenol red exhibits weak estrogen-like activity that can influence hormone-responsive cells or interfere with assays sensitive to estrogenic compounds. It may also cause background signals in techniques like flow cytometry or fluorescence-based assays. For these situations, phenol red-free formulations are preferred.
2. Sodium Bicarbonate–CO₂ Buffering System
Sodium bicarbonate (NaHCO₃) is a natural, non-toxic buffer commonly used in cell culture media. It requires a 5% CO₂ incubator for buffering, as CO₂ dissolves in the medium, forming carbonic acid, which interacts with bicarbonate ions to stabilise pH.
The amount of NaHCO₃ in the medium dictates the amount of CO₂ that should be used to maintain the pH. Physiological pH is generally considered to be in the range of 7.2 to 7.4 for normal tissues, but some disease states (such as cancer) may require culture at a lower pH. Eagle’s Minimal Essential Medium (EMEM) supplemented with Earle’s Balanced Salt Solution (EBSS) and most other cell culture media typically contains 26mM sodium bicarbonate, whereas DMEM has a higher concentration of 44mM.
3. HEPES Buffer – For pH Stability Outside CO₂ Incubators
HEPES (4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid) is a zwitterionic organic buffer widely used in cell culture to maintain stable pH, especially in environments where CO₂ levels may fluctuate. HEPES is particularly effective within a pH range of 6.8 to 8.2, making it suitable for a variety of mammalian cell lines.
Why Use HEPES?
Strong Buffering Capacity: HEPES offers more stable pH control than sodium bicarbonate, especially in environments where CO₂ concentrations are unstable.
CO₂-Independent Buffering: Unlike bicarbonate, HEPES doesn’t require CO₂ to function, making it ideal for experiments conducted outside standard incubators (e.g., live imaging, flow cytometry, and molecular biology assays).
Minimal pH Drift: HEPES helps prevent rapid pH shifts when cells are exposed to ambient air, improving consistency during handling outside the incubator.
Biowest Basal Media – Flexible Formulations for Every Cell Culture Need
Biowest offers a broad range of basal media formulations tailored to the diverse needs of researchers working with various cell types and experimental conditions. Whether culturing robust immortalised lines, sensitive primary cells, or stem cells, Biowest provides ready-to-use media with or without HEPES and sodium bicarbonate to support optimal pH control and buffering.
Standard Options Include:
With Sodium Bicarbonate: Ideal for use in 5% CO₂ incubators, supporting classic bicarbonate buffering systems.
With HEPES Buffer: For experiments requiring stable pH outside CO₂ incubators, such as live imaging, flow cytometry, or field sampling.
Without HEPES/Bicarbonate: Offers flexibility to customize buffering systems based on specific experimental needs.
7. Endotoxin Levels
Endotoxins, also known as lipopolysaccharides (LPS), are components of the outer membrane of Gram-negative bacteria. These molecules can be released in small amounts during bacterial growth, and in larger quantities upon bacterial lysis. Even trace levels of endotoxins can compromise the health of cultured cells, particularly in sensitive applications such as stem cell expansion, immunotherapy development, bioproduction, or modeling inflammatory diseases. Their effects are often subtle but can lead to erratic, irreproducible, or misleading experimental results.
How Endotoxins Affect Cell Culture Outcomes
Even at low concentrations, endotoxins can:
Stimulate cytokine and chemokine secretion (especially in immune-related cells)
Activate inflammatory signalling and oxidative stress pathways
Disrupt normal cell proliferation, differentiation, and morphology
Impair protein expression in recombinant systems
Reduce cell viability or induce apoptosis—particularly in primary or stem cell cultures
Cell types especially vulnerable to endotoxins include:
Immune cells: macrophages, dendritic cells, PBMCs
Stem cells: MSCs, ESCs, iPSCs
Cells used in transfection, viral vector production, or therapeutic protein manufacturing
Common Sources of Endotoxin Contamination
Endotoxins can enter your culture system through various routes:
Water used for buffer or reagent preparation
Commercially sourced media, sera, and supplements
Contaminated labware (glass or plastic)
Improperly sterilised or reused culture vessels
Exposure to air or poor aseptic techniques
Steps to Avoid Endotoxin-Induced Cell Culture Problems
Use Endotoxin-Free or Ultrapure Water – Water used for preparing buffers, washing glassware, or media dilution must be of high purity (e.g., Type I ultrapure water) and certified endotoxin-free.
Choose Premium, Low-Endotoxin FBS – FBS is a common source of endotoxins. Select lots that are certified by the supplier for low endotoxin levels—ideally below 10 EU/mL for general work and <1 EU/mL for sensitive applications. Biowest FBS is LAL-tested and supplied with batch-specific certificates.
Confirm Endotoxin Testing of Media and Additives – Ensure that basal media, supplements (e.g., growth factors, antibiotics), and buffers are manufactured under strict quality control and are tested for endotoxin contamination.
Follow Proper Autoclaving Procedures for Glassware – Residual endotoxins are heat-stable, so ensure that glassware is not just sterilised but also depyrogenated when necessary—typically by dry-heat sterilisation at ≥250°C for 30 minutes.
Use Certified Endotoxin-Free Plasticware – Opt for single-use plasticware (e.g., pipette tips, tubes, flasks) that is certified as endotoxin-free by the manufacturer. Avoid reusing or washing labware that may have been exposed to environmental contaminants.
Minimise Exposure to Environmental Contaminants – Work quickly in sterile environments. Open culture vessels only under a laminar flow hood and reduce handling time outside incubators to minimise the risk of environmental endotoxin exposure.
Biowest Quality Assurance
Biowest provides FBS and basal media that are LAL-tested and supplied with lot-specific certificates of analysis for endotoxin levels, ensuring consistency and reliability across experiments. Their stringent quality control processes support reproducible, high-performance outcomes—even in the most sensitive applications.
Biowest’s Ultra-low-Endotoxin FBS is specifically designed to meet the rigorous demands of sensitive cell culture systems. Each lot is carefully formulated and thoroughly tested to comply with strict endotoxin thresholds, making it an ideal choice for culturing primary cells, stem cells, and cells destined for clinical or biopharmaceutical applications. With Biowest, you can be confident your cells are growing in an environment that supports both viability and integrity.
Conclusion
Cell culture is as much a science of numbers as it is of technique. Mastering key quantitative parameters—like passage number, seeding density, and confluency—can mean the difference between reproducible results and experimental failure. Equally important are less visible but critical metrics such as viability, mycoplasma testing frequency, culture medium pH, and endotoxin levels. Together, these seven numbers form the foundation of a robust, reliable cell culture system. By staying vigilant and informed about these values, researchers can improve consistency, protect cell health, and ensure meaningful outcomes—whether working with standard cell lines or advancing sensitive clinical applications.
Special Offer: Atlantis Bioscience x Biowest – Buy 10, Get 1 FREE!
Don’t miss this exclusive promotion from Atlantis Bioscience and Biowest! For a limited time, when you purchase any 10 bottles of Biowest’s premium cell culture media or reagents, you’ll receive the 11th bottle free—automatically applied to the lowest-value item in your order. This is the perfect opportunity to stock up on high-quality essentials for your research while saving more.
Discover how cell therapy stability requires multi-parametric assays beyond viability to preserve function, phenotype, and safety during storage and thaw.
HOW CAN WE HELP YOU?Our specialists are to help you find the best product for your application. We will be happy to help you find the right product for the job.